
基因治疗一直是亨廷顿舞蹈病(HD)患者和家属的希望和寄托,本月初,关于HD的基因治疗又取得突破性的研究进展啦,让我们一起来回顾一下吧!
HD是由于亨廷顿蛋白(HTT)基因中的CAG三核苷酸重复序列异常扩增引起的。该基因的异常扩增可以引起其编码的蛋白产物长度变长,堆积在细胞内产生细胞毒性,引起神经细胞的功能失调和死亡,从而导致疾病的发生。

图1:HTT基因CAG异常重复致病图示。
因此,想要根治该疾病,应该从最源头的基因层面,阻止编码异常蛋白的过程,减少有害蛋白的产生。
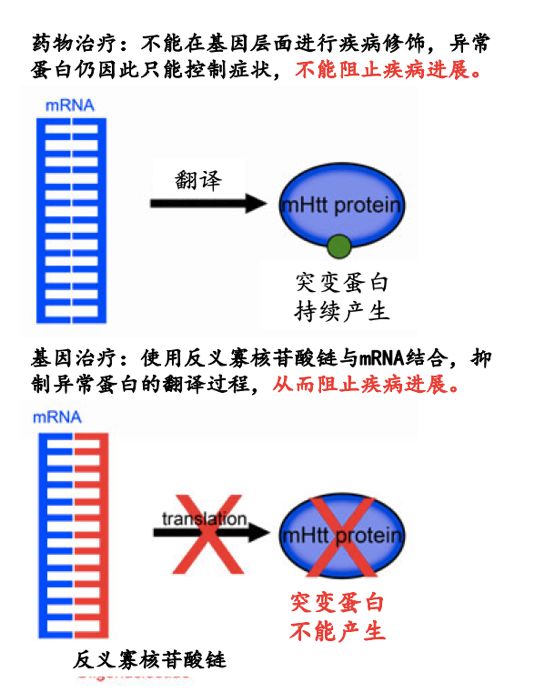
IONIS-HTTRx(也称为ISIS 443139或RG6042;以下称为HTTRx)是第二代2-甲氧基乙基)反义寡核苷酸,可降低正常和异常HTT信使RNA(mRNA)的浓度,抑制正常和突变HTT蛋白的产生。
在HD动物模型中已经发现HTT-Rx可以改善疾病表型。其他用于抑制HTT表达的替代方法的实验在HD的动物模型中产生了类似的效果,表明抑制HTT表达、降低突变HTT的浓度可作为潜在可行的疾病修饰治疗策略。2019年5月6号发表与新英格兰杂志的一篇临床试验报告了将HTTRx用于治疗早期HD成人患者的1/2A阶段临床试验的结果。

本研究纳入的研究对象为年龄在25至65岁之间、基因确诊的早期HD患者。早期HD患者的定义为使用统一亨廷顿氏病评定量表(UHDRS)总功能评分(TFC)的1期患者(分为11至13,职业、财务、家务、日常生活活动和护理水平几乎没有功能障碍)。
该试验于2015年8月至2017年11月在英国、德国和加拿大的9个中心进行。采用集中式自动随机化系统,以3:1的比例分配患者,在5个递增的剂量组(10mg,30mg,60mg,90mg或120mg)分别接受HTTRx或安慰剂治疗。 每位患者每隔4周接受1次鞘内注射HTTRx或安慰剂(人工脑脊髓液为载体),共4次;随后,进行为期4个月的随访,在此期间不给予试验药物。主要终点指标是HTTRx治疗的安全性。
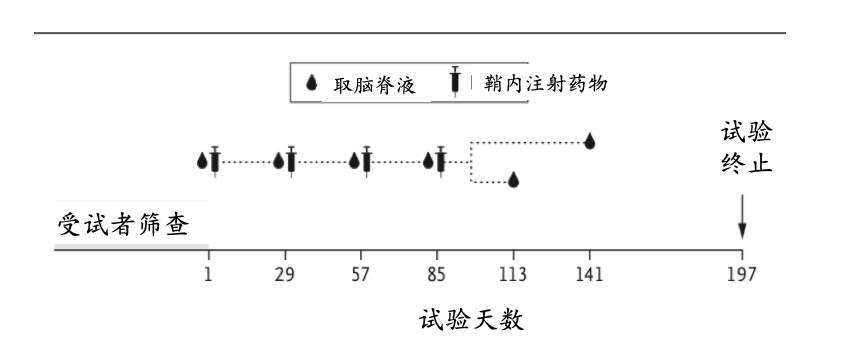
图3:试验流程简图。
从2015年8月至2017年4月,共筛选了52名符合纳入标准的早期HD患者,并根据方案对46名患者进行了随机分组。所有患者均接受所有计划剂量的指定试剂(HTTRx或安慰剂),所有接受随机化的患者均按照方案完成试验。
接受HTTRx的患者和接受安慰剂的患者的不良事件发生率相似(表1)。 98%的患者报告了不良事件;所有事件的严重程度均为轻度(83%)或中度(17%)。接受HTTRx的患者中最常报告的不良事件是手术后疼痛和硬脑膜穿刺后头痛,这种情况腰穿后的发生率为10%,并且与试验持续时间和剂量没有明显的关系。
在接受HTTRx的患者中,在最后一次28天的剂量后采样点,CSF中突变HTT浓度呈剂量依赖性降低(在10-mg,30-mg,60-mg,90-mg和120-mg剂量组平均百分比从基线变化在-20%, -25%, -28%,-42%和-38%),个体患者最多下降率为-63% (在一个接受120mg治疗的患者中)。在接受安慰剂治疗的患者中,CSF中突变HTT蛋白浓度相对于基线的平均百分比变化增加了10%。
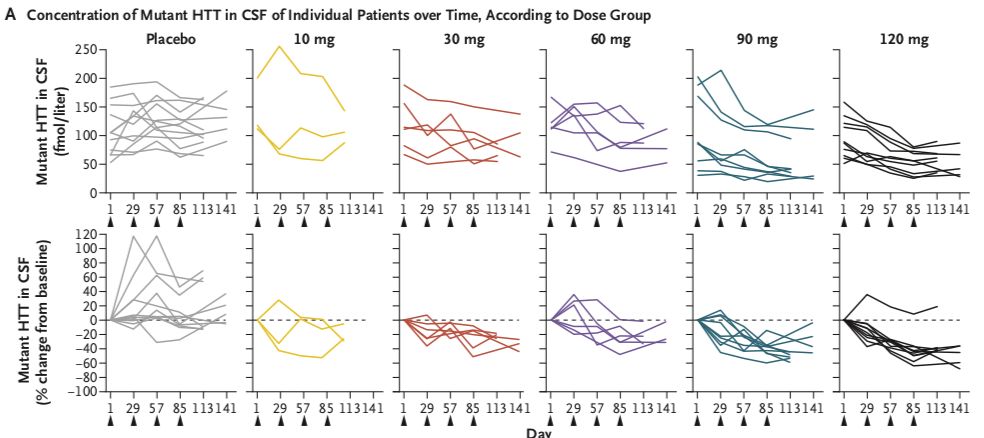
图4:不同剂量组患者脑脊液中突变HTT蛋白浓度的变化情况。
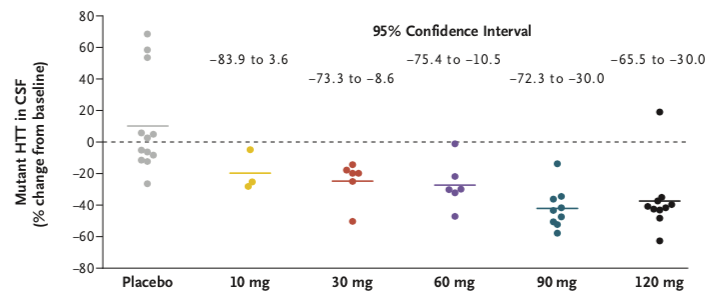
图5:不同剂量组患者脑脊液中突变蛋白的平均下降率。
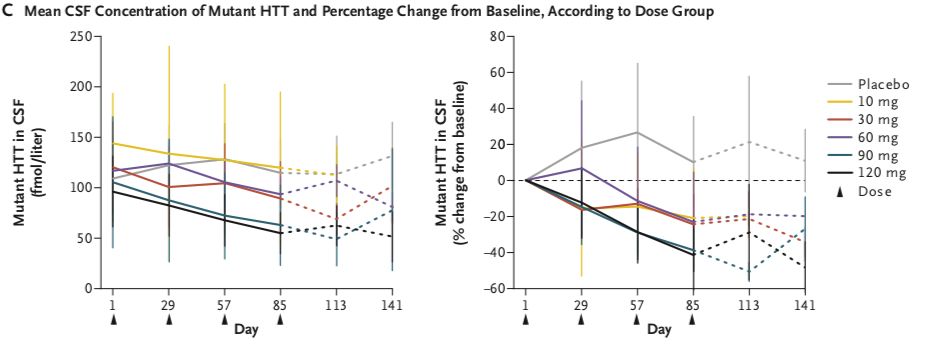
对早期HD患者鞘内施用HTTRx并未出现严重的不良事件,表明其安全性较好。研究结果也发现鞘内注射HTTRx后,脑脊液中突变HTT蛋白浓度呈剂量依赖性降低。
总的来说,该研究证实了在神经退行性疾病患者中鞘内注射反义寡核苷酸可降低蛋白质的表达,对于以后的临床试验具有重要的基石性作用。
Tabrizi SJ, et al. Targeting Huntingtin Expression in Patients with Huntington’s Disease. N Engl J Med. 2019 May 6. doi: 10.1056/NEJMoa1900907. [Epub ahead of print]
翻译:顾孝静
设计编辑:曹蓓
审校:商慧芳

